

上篇 | 生物仿制药业2020年度回顾报告

生物仿制药2020年度回顾
01. 2020 年生物仿制药的批准和上市情况
02. 生物仿制药最新监管资讯
03. 与生物制品和生物仿制药相关的立法
04. BPCIA 诉讼
05. 反垄断诉讼
06. 专利审判与上诉委员会 (PTAB) 受理的授权后挑战
07. 结语
2020 年生物仿制药的批准和上市情况
自 2015 年 FDA 批准首个生物仿制药——山德士的 Zarxio®(非格司亭-sndz)以来,美国的生物仿制药年批准量在 2020 年首次出现下降。2019 年,FDA 批准了十种生物仿制药,创造了新记录。但在2020 年,FDA 仅批准了三种新的生物仿制药:Mylan 的 Hulio®,一种 Humira®(阿达木单抗)生物仿制药;辉瑞的 Nyvepria™,一种 Neulasta®(培非格司亭)生物仿制药;以及 Amgen 的 Riabni™,一种 Rituxan®(利妥昔单抗)生物仿制药。这三种新获批的产品都不是各自参比产品的第一种生物仿制药。
此外,2020 年上市的新生物仿制药也较少,只有五种,而在 2019 年有七种。2020 年新上市的生物仿制药包括:
一种 Rituxan®(利妥昔单抗)生物仿制药,即辉瑞的 Ruxience®;
一种 Remicade®(英夫利昔)生物仿制药,即 Amgen 的 Avsola™;以及
三种 Herceptin®(曲妥珠单抗)生物仿制药,即 Celltrion 的 Herzuma®、Samsung Bioepis 的 Ontruzant® 以及辉瑞的 Trazimera™。
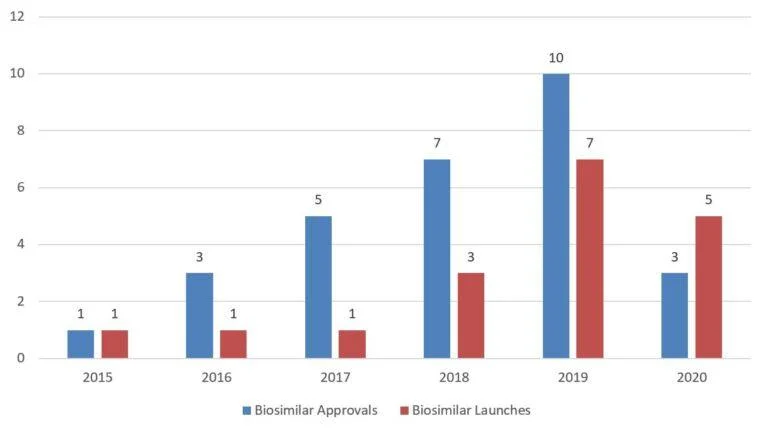
下方图 1 提供了 2015 年至 2020 年间的FDA 批准与生物仿制产品上市趋势概要。请注意,FDA 尚未将任何生物仿制药指定为其参比产品的可互换制品。
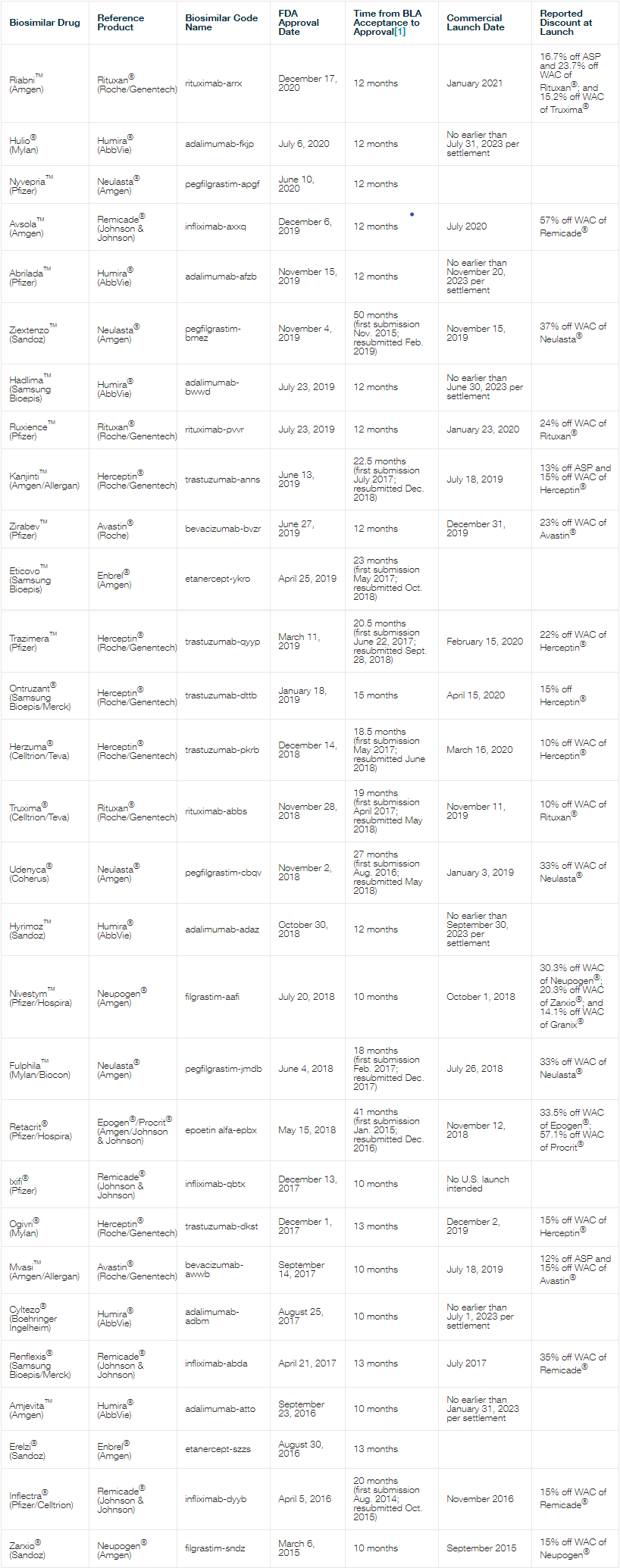
下方表格汇总了已获批和部分待批准的生物仿制药的生物制品许可申请 (BLA) 公开信息,并展现了生物仿制药业的其他趋势。
表 1 汇总了截至 2020 年获批生物仿制药的相关信息。FDA 批准了 29 种生物仿制药,这些生物仿制药分别对应于九种不同的参比产品。虽然2020 年 FDA 批准的生物仿制药总量与过去几年相比有所下降,但是这些生物仿制药的审查周期(从FDA 接受 BLA 到批准通常需要 12 个月)看起来没有受到新冠疫情的影响。到 2020 年底,在 29 种获得批准的生物仿制药中,已有 18 种在美国上市。
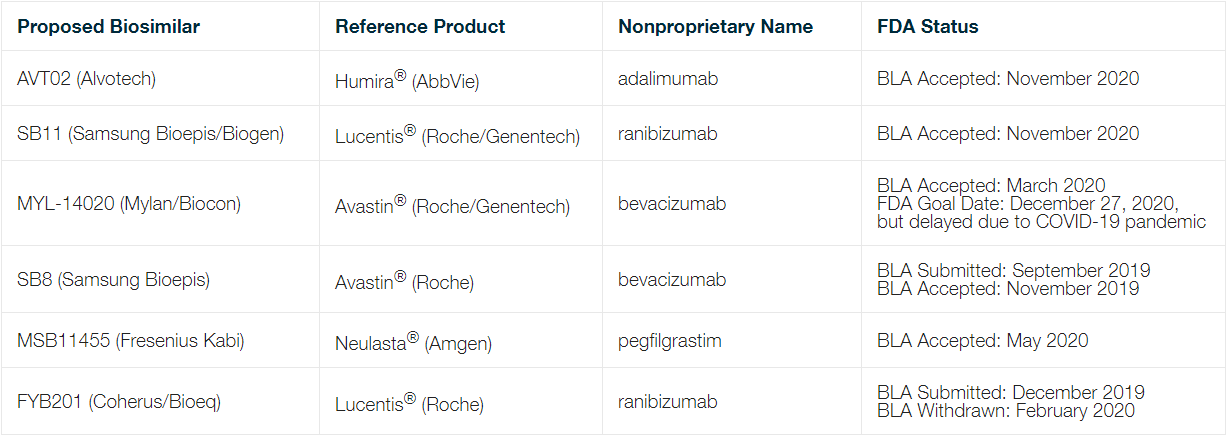
此外,FDA 可能即将批准首个胰岛素生物仿制药。2020 年 8 月 31 日,Biocon Biologics 和 Mylan 宣布推出 Semglee™(甘精胰岛素注射液),该产品于 2020 年 6 月通过根据《联邦食品、药品和化妆品法》(FD&C) 提交的新药申请 (NDA) 获批为赛诺菲 Lantus®的等效产品。2020 年 3 月 23 日,Mylan 针对Semglee™ 的 NDA 被“视为”BLA,Mylan 宣布已向FDA 提交“所有必要文件”,申请将 Semglee™ 批准为生物仿制药和可互换生物仿制产品。
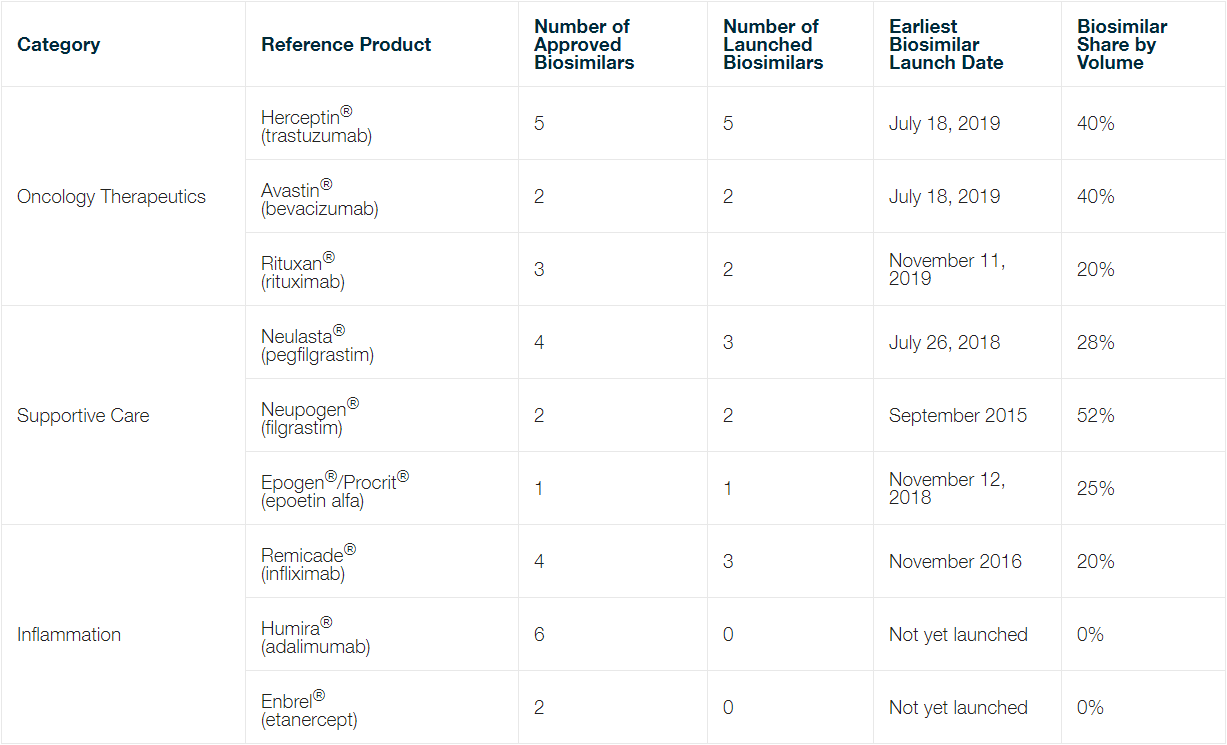
2020 年,生物仿制药的市场吸纳量总体呈上升趋势。多个来源[3]均报告,支持性护理生物制品(Neulasta®、Neupogen® 和 Epogen®)的生物仿制药的当前市场吸纳量约占 25%-50% 的市场份额。肿瘤治疗药物(Herceptin®、Avastin® 和 Rituxan®)的生物仿制药占据各自市场的 20%-40% 份额。这包括 2019 年下半年推出的三种生物仿制药 Mvasi™(贝伐单抗)、Kanjinti™(曲妥珠单抗)和 Truxima®(利妥昔单抗),它们迅速进入了参比产品的市场。
不过,抗 TNFα 抗体(包括 Remicade®、Humira®和 Enbrel®)的生物仿制药仍难以进入市场和取得重要立足点[4]。截至 2020 年 12 月,只有 Remicade® 面临着生物仿制药竞争,并且,尽管 Remicade® 的生物仿制药早于2016 年就已上市,但其仅占据 20% 的市场份额。Humira®和 Enbrel® 的生物仿制药均未上市。按照与艾伯维的和解协议,所有当前获批的Humira® 生物仿制药都须等到 2023 年才可上市。Enbrel®生物仿制药的制造商(山德士和 Samsung Bioepis)卷入与Immunex 的专利诉讼中,并如下文所述,山德士最近在联邦巡回法院的上诉中败诉。
生物仿制药最新监管资讯
2020 年,FDA通过指导文件、公民请愿书和其他机制,持续聚焦生物仿制药。
1)FDA 关注生物仿制药竞争
2020 年2 月,FDA 和联邦贸易委员会 (FTC) 采取措施鼓励生物仿制药竞争,并侧重于监管真实且不具误导性的广告。2020 年 2 月 3 日,FDA和 FTC 发布了《美国食品药品监督管理局和联邦贸易委员会关于合作以促进生物制品市场竞争的联合声明》。该声明提出了四项明确目标:(1)促进生物制品市场的竞争,(2) 制止有碍获取生物制品开发所需样品的行为,(3)采取适当措施来防范有关生物制品的虚假或误导性宣传,以及 (4) 审查涉及生物制品的专利和解协议是否违反反垄断法。
与此同时,FDA 还发布了名为《处方生物参比产品和生物仿制产品的宣传标签和广告注意事项问答》的指南草案。该指南解释称,确定宣传材料是否真实且不具误导性,需要根据“信息的呈现方式”、“所依据数据的类型和质量”、“背景和披露考量”等诸多因素进行“特定于事实”的分析。指南进一步指出,如果宣传材料会使人认为参比产品与生物仿制产品在安全性、纯度或效力方面存在临床上有意义的差异,例如,指出参比产品比其生物仿制药更安全或更有效,或生物仿制药与其参比产品并非高度相似,则宣传材料可能虚假或具有误导性。
差不多在同时,FDA 还回应了辉瑞的公民请愿书,该请愿书初始提交于 2018 年 8 月。在请愿书中,辉瑞提请 FDA 尽快发布指南,“以确保发起人在生物仿制药(包括可互换生物制品)相对于其参比产品的安全性和有效性方面进行真实且不具误导性的宣传”。FDA批准了辉瑞公民请愿书中涉及请求 FDA 发布关于生物制品和生物仿制药宣传标签和广告注意事项的指南草案的部分,但否决了辉瑞关于在指南中纳入具体内容的请求,此等具体内容将由FDA 在考虑公众对指南的意见后确定。
2020 年12 月,Boehringer Ingelheim (BI) 提交了一份公民请愿书,请求FDA 将其对 BPCIA 中就注射用溶液所用“强度”一词的解释更改为“药物总含量”,而不考虑浓度。根据BI 的请愿书,有必要更改解释,以实现以下目的:(1)“确保[FDA] 的解释与 [BPCIA] 的明确含义一致;(2) 防止滥用‘常青化’策略来扼制平价生物仿制药和可互换生物制品竞争;并 (3) 对所有处境相似的注射用生物制品维持公平一致的处理方式”。BI指出,FDA 的当前解释“鼓励或至少允许品牌发起人将不明显的浓度变化用作反竞争策略”。BI以 Humira®(阿达木单抗)为例。BI和其他生物仿制药制造商已有浓度为 50 mg/mL 的获批Humira® 生物仿制药,但是自 2018 年7 月以来,Humira® 仍以 100mg/mL 的浓度进行销售,并且根据 FDA 的当前解释,目前没有获批的阿达木单抗生物仿制药可视为这一高浓度产品的生物仿制药或可互换生物制品。FDA确认已收到 BI 的请愿书,但是尚未提供任何实质性反馈。
2)FDA 提供有关生物仿制性和可互换性的附加指南
2020 年2 月,FDA 发布了名为《生物仿制药和可互换生物仿制药:根据参比产品已获许可的部分使用条件授予许可》的指南草案。在该指南草案中,FDA 确认“生物仿制药或可互换生物仿制药只能根据参比产品已获许可的使用条件获得许可”。但是 FDA 承认“存在各种不同情况”,例如罕见病药物的专营保护期和专利保护,这可能导致申请人为较少的适应症寻求许可。指南草案提供了有关如何计划为部分而非全部适应症寻求批准的建议。
2020 年11 月,FDA 发布了名为《生物仿制性和可互换性:有关生物仿制药开发和 BPCI 法案的附加草案问答》的新指南草案。除其他内容外,这份新指南草案还说明了 FDA 目前关于向 FDA 寻求审查生物仿制药和可互换生物仿制药 BLA 及可互换生物制品标签的看法。例如,指南草案指出,FDA 计划将根据第351(k) 条提交的新 BLA 视为新生物仿制药申请(如指明,也包括可互换生物制品),除非申请信明确指出申请人仅将其作为可互换生物制品来寻求许可。该指南还提供了用于在标签上指明可互换性的示范性表述,并阐明,许多关于生物仿制药标签的指南也适用于可互换生物制品标签。例如,指南草案指出,标签中不应包含支持可互换性的临床研究(支持生物仿制性的临床研究也是如此)。
3)将以往列为“药品”的胰岛素和其他生物制品转变为作为生物制品接受监管
2020 年3 月 23 日,BPCIA 的“视为许可”规定开始生效,改变了胰岛素和某些其他生物制品的审批方式。通常,“药品”和“生物制品”需遵循不同的法律法规。药品需按新药申请 (NDA) 遵循 FD&C 法案,而生物制品则受 PHS 法案的监管。以往,一部分生物制品(包括胰岛素和人类生长激素)根据 FD&C 法案作为药品寻求批准,而不是视为PHS 法案项下的生物制品。因此,申请人不能根据 BPCIA 来为其生物仿制药或可互换生物制品寻求批准。但是,自2020 年 3 月 23 日发生转变以来,所有以往根据FD&C 法案寻求批准的“生物制品”申请均被“视为”根据 PHS 法案获得批准,公司现可申请这些生物制品的生物仿制药和可互换生物制品。
FDA 为此转变开展了准备工作,包括发布相关指南,并制定了一项最终规则,以确定转变范围所含“生物制品”的定义。根据该规则,FDA 按照法规中的定义调整了“生物制品”的监管定义,法规指出“生物制品”包括“蛋白质”。例如,FDA 解释称,“蛋白质”术语的含义是“具有指定的既定序列且大小超过 40 个氨基酸的任何氨基酸聚合物”。
如上文所述,业内公司已在利用这一新的类别指定。具体而言,Mylan 已宣布向 FDA 提交了“所有必要文件”,以使 Semglee™(一种胰岛素产品)作为可互换生物仿制药生物制品获得批准。
4)FDA 更新紫皮书
去年,FDA 已将“紫皮书”转变为可搜索的公开在线数据库。新的可搜索数据库可通过https://purplebooksearch.fda.gov/ 访问,目前其中包含有关“所有获得 FDA 许可(批准)的由药品评价和研究中心(CDER) 监管的生物制品,包括获得许可的生物仿制药和可互换生物制品及其参比产品”以及“所有获得 FDA 许可的由 CDER 监管的过敏原、细胞和基因治疗、血液和疫苗制品”的信息。紫皮书目前包含批准日期、获批BLA 类型以及是否有获批生物仿制药或可互换生物制品等方面的信息。尽管紫皮书不含任何专利信息,不过根据近期立法,这将在未来发生改变(参见下文)。
5)FDA 应对全球新冠疫情
为应对全球新冠疫情,FDA 采取了多项措施,包括推迟检查。这至少阻碍了一种预期的新生物仿制药,即 Biocon 和Mylan 的 Avastin® 生物仿制药MYL-14020。据报道,FDA 推迟了对该生物仿制药的最终决定(原本预计在2020 年底作出),这是由于新冠疫情导致的出行限制而使 FDA 无法对制造设施进行检查。
FDA 还发布了一系列指导文件,帮助公司应对疫情带来的新挑战。例如,2020 年 6 月,FDA 发布了《药品和生物制品制造业员工应对新冠病毒感染的良好生产规范考虑因素》,这份指导文件就预防污染的制造控制措施、与药品安全或质量有关的 SARS-CoV-2 风险评估以及制造运营的连续性,为药品和生物制品制造商提供建议。2020年 8 月,FDA 发布了名为《新冠疫情公共卫生紧急事件期间的制造、供应链、药品和生物制品检查问答》的指南,其中除了其他内容外,还阐明 FDA 如何处理制造场所检查,并针对被视为“关键任务”的检查提供见解。2020年 9 月,FDA 发布了名为《新冠疫情公共卫生紧急事件期间恢复正常药品和生物制品制造运营》的文件,其中说明新冠疫情期间“如何评估对必须延迟、减少或以其他方式修改的 [现行良好生产规范 (CGMP)] 活动的补救并确定其优先顺序”,“以维持生产和药品供应”。2020 年 12月,FDA 发布了名为《新冠疫情公共卫生紧急事件期间医疗产品临床试验的开展》的指南,为新冠疫情期间开展临床试验的安全性和良好药品临床试验规范 (GCP) 依从性提供指导意见。
与生物制品和生物仿制药相关的立法
2020 年,联邦和州级的立法机构审议了与生物制品和生物仿制药相关的多项立法提案,这些提案旨在提高患者可及性、鼓励商业化和降低成本。
1)联邦立法
2020 年12 月 27 日,美国总统签署了第二份新冠疫情刺激法案,其中包括对BPCIA 的修订和对紫皮书的更新,后者中列出了获得 FDA 许可的生物制品(包括获得许可的生物仿制药和可互换生物制品及其参比产品)。这些修订使紫皮书与针对小分子药品的类似橙皮书更接近了。具体而言,根据新的修订,参比产品发起人(RPS) 需在专利作为“专利舞蹈”组成部分首次提供给生物仿制药申请人后 30 天内,向 FDA 提供任何专利清单的副本和专利到期日期(根据《美国法典》第42 编第 262(l)(3)(A)或 (l)(7)条)。随后,FDA 需将此专利信息以及关于各获批生物制品的以下信息加入公共“可搜索电子”数据库(即紫皮书):非专有名称、许可日期和申请号、许可和营销状态,以及专营保护期。
2020 年还提出了多项针对生物仿制药的立法提案,这些提案主要关注提高生物仿制药的可及性,以及降低生物类药品(包括胰岛素)的患者成本。虽然这项立法未在第116 届美国国会任期颁布,但它提供了对这些问题的见解,而这些问题可能是未来立法的重点。
例如,2020 年 3 月,多位代表 Tony Cárdenas(D-CA)、Richard Hudson (R-NC)、BrianFitzpatrick (R-PA) 和 Angie Craig (D-MN) 提出了《2020 年提高生物仿制药可及性法案》(H.R.6179)。2020 年 7 月,议员John Cornyn (R-TX) 和 Michael Bennet (D-CO) 提出了同名的相伴法案 (S. 4134)。这些法案将指导医疗保险和医疗补助服务中心(CMS) 实施“共享节省”模式,通过按一定比例向医疗服务提供商提供其实现的任何净节省金额,鼓励医生开具更低价格的生物仿制药,从而减少患者的医疗支出。
同样在 2020 年 3 月,议员 Martha McSally (R-AZ) 和 Doug Jones (D-AL) 提出了《2020 年生物仿制药可及性法案》(S. 3466)。该法案规定,在生物仿制药上市后的初始5 年间,免除医疗保险 B 部分计划受益人购买生物仿制产品的所有自付费用。这是2019 年 10 月在美国众议院提出的 H.R.4597 的相伴法案。
2020 年9 月,Glenn Grothman (R-WI) 代表提出了一项新法案,即《2020年生物仿制胰岛素可及性法案》(H.R.8190),该法案将允许生物仿制胰岛素自动获得其参比产品的可互换生物制品批准。
2)州立法
许多州已颁布了针对生物仿制药(特别是针对未来的可互换生物制品)的法律,而加利福尼亚州还颁布了若干额外的特殊法律。具体而言,出于降低药品价格的明确目标,加利福尼亚州最近颁布了两项医疗保健法律 AB 824 和 SB 852。
2020 年1 月 1 日,824 号加州众议会法案(AB 824) 生效。这项史无前例的法律由加州州长 Gavin Newsom 于 2019 年 10 月签署,旨在在品牌药制药公司或RPS 向仿制药或生物仿制产品制造商支付费用,以拖延或阻止较低价药品进入市场时,用于监管此等反竞争专利和解(有时称为“有偿延迟”协议)。这项加州新法律最显著的特点是,如果利用仿制药或生物仿制药获得“任何有价物”,则假定此等和解是反竞争和非法的。这项假定违背了最高法院在2013 年 FTC诉 Actavis 案中就分析专利和解合法性所提出的传统框架,根据该框架,应由反垄断原告承担举证责任。此外,AB824 还规定,加利福尼亚州可对违法行为处以民事罚款,“金额最高为可合理归因于违法行为的当事方所获价值的三倍”或2,000 万美元,以两者中的金额较高者为准。美国无障碍药物协会 (AAM) 质疑 AB 824 违反宪法,且应受限于联邦法律优先适用原则。
2020 年9 月,州长 Newsom 签署了《2020 年加利福尼亚州平价药品生产法》(SB 852),该法律允许加州卫生与公共服务部与药品制造商和供应商签订合同,以生产和分销其自有品牌的生物仿制药、生物仿制胰岛素和仿制药。在宣布该新法律的声明中,州长 Newsom 指出:
医疗成本太高了。我们的法案将有助于为仿制药市场注入竞争力,将定价权从大型制药公司交还给消费者……加州正在利用我们的市场力量和道德力量,要求处方药实现更公平的价格。我很自豪能签署这项法律,确认我们在打破市场壁垒、提供平价处方药方面的开创性领导地位。
声明:中文翻译仅供读者参考,请以英文原文为准
知产前沿新媒体将于明天推送本报告另外四个部分,敬请关注:




